
Pharmacologic Terms and Concepts
Byron C. Yoburn
College of Pharmacy & Allied Health Professions
St. John's University
Queens, NY 11439
Pharmacologic Terms and Concepts Byron C. Yoburn College of Pharmacy & Allied Health Professions St. John's University Queens, NY 11439
Home - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - Comments or specific questions??
Select a letter from the drop down list to reach terms beginning with that letter
Action potential
Adenylate Cyclase: see adenylyl cyclase
Angina (syn. Angina Pectoris): Paroxysmal pain in the chest usually assumed to be caused by increased demand for oxygen by the heart. Usually classified as typical angina or variant angina.
Angiotensin Converting Enzyme: See ACE inhibitor.
Antagonism: Antagonism refers to inhibition or blockade of an agonist induced response. There are several forms of antagonism including: chemical, physiologic and pharmacologic. In addition antagonism can be described as competitve, noncompetitive, reversible, irreversible, surmountable or insurmountable.
Antagonist: An antagonist blocks, inhibits or interferes with the effects of an agonist or neurotransmitter. (see Neutral Antagonist)
Anti-anxiety: See anxiolytic
Antiarryhthmics: Drugs that are employed to alter abnormal rhythms of the heart. Abnormal rhythms generally consist of disorders of a disturbance of cardiac rate, rhythm, or both. The most common scheme for classifying the antiarrythmics is that of Williams which categorizes drugs into a Class system:
Class 1: block voltage-sensitive Na+ channels (e.g., lidocaine),
Class II: ß-adrenoreceptor antagonists
Class III: prolong the action potential and consequently effectively prolong refractory period via block of K+ channels or increasing inward current via Na+ or Ca++ channels (e.g., Bretylium, Amiodarone, Sotalol, Quinidine, Ibutilide, dofetilide
Class IV: Ca++ channel antagonists, block voltage-sensitive Ca++ channels. Slow conduction and increase refractory period.
There is also a group of miscellaneous antiarrythmics that are not classified and have various mechanisms and applications:
Digoxin (Atrial fibrillation)
Muscarinic antagonists (Sinus bradycardia)
Adrenoreceptor agonists (Cardiac arrest)
Adenosine (supraventricular arrythmia)
Calcium salts (Ventricular arrhythmia secondary to hyperkalemia)
Magnesium salts (Ventricular arrhythmia)
Antibiotic:
Anticholinergic: A drug that acts to block, inhibit or antagonize the actions of acetylcholine or other cholinergic agonists. Since the predominant effects of the parasymapthetic nervous system are mediated by acetylcholine, the term 'anticholinergic effects' is often used to imply an inhibitory action in the parasympathetic nervous system.
Anticonflict effect: The conflict procedure is a behavioral test that can be used to evaluate drugs for potential antianxiety activity. Animals are trained to respond on a lever for access to some positive reinforcer such as food or water. The availability of reinforcers is programmed according to a schedule of reinforcement, such that every response does not yield a reinforcer. For example, reinforcers are delivered on the average of once per minute; i.e. the first response after an average of 1min has elapsed will be reinforce. The actual time intervals will vary but the mean interval will be 1min. This schedule generates a high rate of responding due to the inherent unpredictability concerning the time to the next reinforcer. Once responding has been stabilized, then responses are punished by the addition of a brief electric shock following each response. Under these circumstances responding is suppressed. The animals are thus in conflict, since responding yields both punishers and reinforcers. When animals are administered antianxiety compounds such as diazepam, punished responding increases (is released) and this is considered to be an anticonflict effect. Syn. Geller Seifter procedure..
Anticonvulsant: A drug class that has its major actions on convulsions or seizures. Anticonvulsants can act prophylactically or acutely.
Antihypertensives:
antipsychotic: A drug class that acts to reduce the core symptoms of psychosis, most usually schizophrenia. The prototype drug group are the phenothiazines (e.g. chlorpromazine). Syn. neuroleptic, antischizophrenic
Antisense:
Antivirals:
Anxiolytic: A drug which reduces anxiety. Syn. Antianxiety agent. e.g. diazepam.
Auto receptor: A receptor that is designed to be sensitive to the neurotransmitter that is released by the nerve terminal upon which the auto-receptor resides. It is usually considered that the function of an autoreceptor is to modulate the release of neurotransmitter at the nerve terminal. Activity at an autoreceptor may also regulate the synthesis of transmitter. (Click Here)
Autonomic nervous system: This branch of the nervous system is composed of the parasympathetic and sympathetic nervous systems. Many drug side effects are mediated by the ANS and as such it is important to understand the major actions of each branch of this system.
Availability: The completeness of absorption determines the availability. The total amount available is the percent of the total drug administered that is actually absorbed. This can also be expressed as fractional availability.
Binding Site: The portion of the receptor to which a ligand (neurotransmitter, drug) actually adheres to. Binding can be based upon weak and/or strong bonds that are formed between the ligand and the binding site. Occupancy of the binding site can lead to a chain of events that culminate in an effect. However, occupancy does not imply an effect since antagonists can bind but produce no direct effect as a result of occupancy.
Bioavailability: The amount of administered drug that is actually therapeutically available to the animal or patient. Bioavailability implies that the amount of drug that is bioavailable has access to its site of action.
Biotransformation: Factors that act to convert a drug to another form. Typically, in the process of drug elimination drugs are biotransformed to inactive, more polar, water soluble products that can be more easily excreted in the urine. However, the products of biotransformation can also be active compounds in themselves, or may be toxic. The major organ of biotransformation is the liver
Bioequivalence: Drugs that are bioequivalent provide the same amount of active drug to the body. This term is used when comparing the same drug manufactured by two companies, or 2 different preparations (e.g. oral vs. im).
Bmax: This term refers to the density of binding sites as determined by receptor binding assays. The Bmax is typically quantified in moles (pico- or femto) per mg (of protein or wet weight). Using a ligand that binds to only one binding site the Bmax can be estimated as the x-intercept of the line of best fit using scatchard analysis. When a ligand binds to more than one site, simple line fitting does not accurately estimate the densities of the binding sites.
Bound: This term is used to refer to the binding of a drug or other compound or element to another molecule or element. Typically, the "bound" fraction of a drug has certain characteristics. For example, protein binding of a drug limits the amount of free drug available to induce an effect. Receptor binding of a drug can elicit a chain of events leading to an effect. Binding to an enzyme may increase or decrease enzyme activity. In most cases, the fraction bound to a receptor or an enzyme is considered to be very small relative to the fraction that is not bound. On the other hand, the amount of drug bound to plasma proteins can be considerable (e.g., 90%)
Calcium: Calcium often exists as a divalent cation. It has important roles in neurotransmission and muscle contraction
cAMP: cyclic Adenosine Monophosphate. A second messenger formed by adenylyl cyclase from ATP. The activity of many intracellular kinases are dependent upon cAMP (hence: cAMP-dependent protein kinase).
Cardiac glycoside: include the digitalis derivatives (digoxin, digitoxin ) derived from the foxglove (Digitalis purpurea, D. lanata) and other sources.. The general effect of these drugs is to increase cardiac output (i.e., positive inotropic effect). Although these drugs have a narrow safety margin, the cardiac glycosides have been a traditional approach to CHF since they correct the failure of contractility. All cardiac glycosides (aka cardenolides) have a similar structure with a steroid nucleus and an unsaturated 5-membered lactone ring at the 17 position and a series of sugars linked to carbon 3 of the nucleus. The cardiac glycosides inhibit Na/K ATPase (pump) which ultimately results in an increase in intracellular [Ca]. Ion relationships for the glycosides (Click Here)
Catecholamine: A group of neurotransmitters that are structurally related by a catechol nucleus. They include norepinephrine, epinephrine and dopamine.
cGMP: cyclic Guanosine Monophosphate. A second messenger formed by guanalyl cyclase from GTP. The activity of some intracellular kinases are dependent upon cGMP (hence: cGMP-dependent protein kinase). Among the roles for cGMP are its contribution to relaxation of vascular smooth muscle .
Cephalosporin: point here
Central nervous system: Refers to neural tissue which is enclosed within cranium and the vertebral column. The central nervous system consists of the brain and the spinal cord.
Chemical Antagonism: Chemical antagonism can be nonreceptor mediated. A common example of chemical antagonism is the scenario in which one drug can bind to and inactivate an agonist, thus making less of the drug available to produce an effect.
Cholinergic: Refers to biological systems that rely on acetylcholine as a neurotransmitter. There are two fundamental neuropharmacologic types of cholinergic neurons: nicotinic and muscarinic. There are three basic anatomic divisions of cholinergic neurons: neuromuscular, ganglionic and post-ganglionic.
Clearance: A fundamental pharmacokinetic term that refers to the ability of the body to eliminate a drug. The term is borrowed from renal physiology and is based on the concepts developed for creatinine clearance. It is defined as: Clearance = (rate of elimination)/ (concentration of drug in biofluid). The biofluid can be blood, plasma or other relevant fluid. Total body clearance is actually the sum of clearance by renal, liver and other mechanisms.
Compartment models (1 & 2): These are pharmacokinetic conceptualizations of the way a drug can localize or distribute in a biological system. In the simplest form, a one compartment model refers to a drug that distributes only to the vascular space. Thus, the drug is confined to a single compartment. If the drug also distributes out of the vascular space (to muscle, CNS, etc.), then this non-vascular distribution can be viewed mathematically as a second compartment. It is also possible to have more than 3 compartments (CNS, fat, muscle can all be theoretically defined separately) but in many cases the clinical utility of the complex kinetic modeling has practical limitations. There are typical signature elimination functions for one and 2 compartment models and they are easily recognized.
Competition Assay: (syn. Displacement Assay) In this type of binding assay, two ligands that bind to the same site are available in the tube. Each ligand 'competes' for the binding site. Typically one of the ligands is radiolabelled (tracer or hot) and its concentration is held constant, while the other ligand is not labeled (cold) and its concentration is varied over a fairly wide range. As the 'cold' ligand's concentration is increased the amount of labeled ligand (trace) on the binding site is decreased. Eventually the amount of labeled ligand on the binding site is separated from that in solution (i.e. separation of bound from free) and the amount of label is counted. Graphs of this data are quantified by examining the IC50, Ki; as well as determining if more than one binding site is present. Curve fitting by nonlinear regression is used to obtain these parameters. (Click Here)
Competitive Antagonist: An antagonist that acts at the same binding site as an agonist. Point Here
Concentration: In pharmacokinetics, the amount of drug (e.g., mg, m g) per unit of physiologic tissue (e.g., ml of blood, mg of fat).
Confidence limits: A range of values around a mean value that provides an estimate of the most likely interval that contains the true mean value. Confidence limits are often expressed as the 95% or 99% interval. The greater the confidence (99% vs. 95%), the larger the interval. Confidence intervals are used since in any given experiment it is likely that our calculated mean value is close to the true value but is unlikely to be an exact estimate. In actuality, the confidence interval may not contain the actual value, but the calculation of the interval allows us to to state with a degree of certainty (e.g., 95%) that the real value is within the interval. Furthermore, when reviewing data in which the confidence limits are broad, this suggests that there was significant variability in the individual observations that were used to calculate the mean.
Conflict procedure: See anticonflict effect.
Cooperativity: A cooperative interaction is one in which the binding of one molecule to a binding site affects the binding of other molecules. Reactions can be positively cooperative or negatively cooperative. A common example of positive cooperativity is the binding of O2 to hemoglobin, in which the binding of one molecule increases the affinity of binding for additional O2. See Hill Coefficient
Constitutive Activity:
Cumulative Dose-Response: A dosing procedure in which subjects are repeatedly administered a drug and tested after each dose until a criterion effect is reached. Typically, in a cumulative dose-response protocol, subjects are injected with an initial dose (i.e., starting dose) of a drug and tested for some criterion effect. Subjects that do not reach the criterion effect are given a second dose (i.e., increment dose) of the same drug and then retested. This dosing procedure is continued until a predetermined percentage of subjects respond; usually 80-100% of subjects. The percent responding at each cumulative dose is plotted against the cumulative dose. As with the standard dose-response protocol, an ED50 and relative potency estimates can be calculated. By employing a cumulative dose-response protocol the number of subjects and drug used can be reduced compared to a standard dose-response protocol in which each subject receives a single dose at a time.
Dependence: Dependence has been historically defined as the appearance of a withdrawal syndrome following discontinuation of administration of a drug (e.g. morphine, secobarbital, alcohol). The appearance of the withdrawal syndrome has been taken as a defining factor in physical dependence. However, more recently, the idea has been expressed that drug dependence may not entail physical dependence and withdrawal. Thus, psychological dependence and terms such as neuroadaptation have found their way into the drug dependence vocabulary. Psychological dependence can occur in the absense of a withdrawal syndrome following termination of dosing. Similarly, physical dependence doed not necessarily imply that psychological dependence has developed. Neuroadaptation implies that chronic drug use will produce changes in the nervous system, but that these changes do necessarily mean that psychological dependence has developed or that a withdrawal syndrome will occur.
Dephosphorylation
Desensitization: There has been some confusion in the pharmacology literature as to the exact meaning of desensitization. In a general sense, desensitization indicates a reduced response to an agonist. This can be quantitated using dose-response analysis in which a shift to the right or a reduction in Emax indicates loss of sensitivity. Desensitization is often used to suggest a reduced response that occurs over a relatively short period of time, with terms such as tolerance reserved for reduced responsiveness developing over longer periods. However, this distinction in terminology is not universal. Theoretically, desensitization could include any mechanism that reduces the effect of an agonist, including: receptor phosphorylation, uncoupling or receptor downregulation. On the other hand, receptor desensitization is used to describe a situation in which there is no change in receptor density, but the signaling properties of the receptor have been reduced. This may be due to uncoupling of the receptor from intracellular signaling molecules (e.g., G-proteins) and/or receptor phosphorylation which might reduce functional characteristics of the receptor. Finally, homologous and heterologous desensitization have been distinguished, where the former refers to desensitization induced by prior activation of that receptor system, and the latter refers to desensitization that is a result of activation of another receptor system.
Diacylglycerol: Click Here
diffusion: the tendency of molecules or ions to distribute themselves equally throughout an environment (liquids, gas). see facilitated diffusion.
Digitalis (Syn. cardiac glycoside): A general term often used to refer to a group of compounds derived from digitalis (e.g. digoxin, digitoxin)or that have actions similar to that of digitalis. These drugs are used in cardiac failure and function by increasing cardiac contractility by indirectly increasing intracellular Ca++ availability.
Displacement Assay: see competition assay.
Disposition: The disposition of a drug refers to the fate of the drug once it has been absorbed. The processes that occur following absorption are distribution and elimination. Thus, disposition of an intramuscularly administered drug following absorption into the circulatory system, will be distribution to its site of action followed by elimination of the drug by biotransformation or excretion.
Distribution: A pharmacokinetic term that refers to the movement of a drug from the site of administration into the various tissues and organ systems of the body. In most cases, distribution is used to describe the passage of a drug out of the vascular system to other compartments of the body.
Diuretics: point here
Dopamine: A member of the catecholamine neurotransmitters. Dihydroxyphenylethylamine.
Dose-response analysis: This is the fundamental metric in pharmacology that relates the dose of a drug to a particular effect. There can be many effects of any drug (e.g., sedation, anesthesia, death) and these effects are graphed as a function of the dose of drug. The relative position of the dose-response function for a given drug provides significant information including its relative potency and maximal effect. Comparison of Log and Normal DR (Click Here)
Downregulation: The downregulation of receptors is usually used to characterize a decrease in receptor number or a reduction in affinity. Either of these cases would require more drug to produce a given effect compared to a normal (nondownregulated) system.
Drug: A chemical entity that affects the processes of a living organisms or system.
Drug Approval: Drugs that are approved for use in the United States have undergone a rigorous examination of pharmacodynamics and pharmacokinetics. There are 3 major phases of testing of a drug in humans before it is made available to the public. In Phase 1, the effects of the drug in a small group of volunteers is determined. Phase 1 is a trial to determine the kinetics of the drug and the general effects it might have, including any toxicity. The dose range that might be used in subsequent Phases is usually determine in Phase 1. In Phase 2, the drug is examined in a group of patients that have the target illness. The potential efficacy of the drug compared to placebo, or a standard treatment drug is determined. The majority of drugs that enter Phase 1 and 2 trials never reach Phase 3 due to toxicity or efficacy concerns. In Phase 3, the drug evaluation is expanded to a larger group of patients. In some cases, many thousands of patients may be enrolled in a Phase 3 trial. The previous Phase 1 and 2 information is used to guide the protocol for establishing the Phase 3 trial. In most cases, Phase 3 trials are conducted under circumstances that are close to that which might occur in actual clinical practice. However, the trial is a controlled trial, with double-blind procedures and other appropriate experimental protocol to insure a reasonable test of the drug. If the Phase 3 results are acceptable, the manufacturer may file an application with the FDA. This application is called a New Drug Application (NDA) and will contain significant information about the drug so that the FDA review panel can assess the safety and efficacy of the drug. If approval is granted, the drug moves to a post approval period, or Phase 4 (postmarketing surveillance). In Phase 4 the safety of the approved drug is monitored and low likelihood adverse effects may become apparent. If the incidence or severity of the adverse effects is sufficient, the drug may be removed from the market. (Click Here)
Drug Discovery: The process by which new chemicals that might have specific biologic activity are developed. The chemicals are examined to see if they are active using some assay or biological marker to determine if they might be useful clinically. The identification of a new chemical for clinical use might be based on modification of a standard drug, random testing of many chemicals or a rational approach that targets a known mechanism of disease.
Drug discrimination: A procedure that allows a subjective drug effect to direct choice behavior. In this paradigm, an animal may be taught to turn left when under the influence of one drug and to turn right (or make no response) when another drug (or no drug) is administered. In this manner, the subjective state produced by drugs serve as discriminative stimuli that set the occasion for a particular response. The drug discrimination paradigm can be used to determine if a new drug has subjective properties similar to an established drug.
EC50:: The concentration of drug that produces a half-maximal effect. The EC50 is calculated from graded dose-effect data.
ED50: The Effective Dose for 50 % of the population. This is the dose of a drug that will produce a criterion effect in half the population. For example, if pain relief is defined as 4hrs of no pain, then the dose that provides 4hrs of relief to half of all patients is the ED50. The ED50 is synonymous with the Median Effective Dose.
Effector: A peripheral tissue (e.g., gland, muscle) that is innervated and responds when a nerve impulse occurs.
Efficacy: This has historically been a measure of the effect produced by a drug. It is simple to distinguish the efficacy of a full agonist and a partial agonist, since the former but not the latter can produce a maximal response; and thus the full agonist has greater efficacy. However, the term efficacy can take on a confusing nature when comparing 2 full agonists. When two full agonists that are capable of producing the same maximal response are compared the notion of efficacy must be expanded and consideration of intrinsic activity and intrinsic efficacy must be discussed. Intrinsic activity (a ) was a term originally proposed by Ariens as the "effect per unit of pharmacon-receptor complex". In the original formulation 0£ a ³ 1, where for a full agonist a =1 and for an antagonist a =0. In this formulation, however, even a full agonist must occupy all receptors in order to produce a maximal response. The necessity for an agonist to occupy all receptors so as to produce a full response has not been confirmed. Reformulations of receptor theory removed this requirement and new terms were added by Stephenson and Furchgott. Stephenson introduced e a term which was referred to as efficacy; Furchgott introduced intrinsic activity denoted by the term e . It is related to Stephenson's efficacy (e) term: e = e/[RT]. The advantage is that intrinsic efficacy is a drug-related term and is independent of tissue factors such as receptor density. e represents a quantal unit of the capacity of a drug to initiate a stimulus from one receptorIncreases in intrinsic efficacy will shift the dose-response curve to the left. Decreases in intrinsic efficacy will shift the dose-response curve to the right. Eventually as intrinsic efficacy decreases the maximal response will also decrease. As intrinsic efficacy approaches zero, response approaches zero.
Eicosanoids: oxygenation products of long chain polyunsaturated fatty acids. They are a very large group of compounds with a broad and profound array of effects. They include: prostaglandins, thromboxanes, leukotrienes. All are C 20 compounds, hence the name; in Greek eikosi is twenty.
Elimination: Drug elimination refers to all the processes that result in the removal of the drug from the body. This includes biotransformation and excretion.
Elimination rate constant: In pharmacokinetics, a simple first order elimination function has the form of:
C=Co e-kt
Where C is the concentration at time t, Co is the concentration when t =0, and k is the elimination rate constant. The elimination rate constant is a factor that has units as reciprocal time (1/sec) and has a negative sign since drug concentrations are falling. The elimination rate constant is useful to know since the half life (t1/2) is related to it:
t1/2 = (0.693/k)
For an example of first order elimination kinetics (Click Here)
Emax: The Emax is the maximal effect produced by a drug. Different drugs may have different Emax's and thus some drugs may be more effective for certain conditions. For example, both acetaminophen and morphine are effective analgesics. However, morphine's Emax is far greater than that of acetaminophen. Emax has been used as a measure of efficacy since in some cases it may be proportional to efficacy.
EPSP: Excitatory postsynaptic potential.
Excretion: A pharmacokinetic term that refers to the irreversible removal of a drug in the unchanged form. Excretion is one of two processes (metabolism, excretion) that account for the elimination of a drug from the body. The kidney is the typical site of drug excretion, although excretion may occur at the liver, lungs or in breast milk.
Facilitated diffusion: also called passive transport. Facilitated diffusion does not require an energy source but does invovle a carrier substance that asists in moving the transported molecule down its concentration gradient (sometimes called carrier-mediated diffusion)
First order kinetics: In many cases, drugs are eliminated according to first order kinetics. This means that the function relating drug concentration following administration to time since administration follows an exponential function. As a consequence, a constant fraction of the drug is eliminated in each equal time period. Thus, if 25% of the drug is removed in first hour, 25% will also be removed in the second hour. However, the actual amount of drug eliminated will vary. So if the drug concentration in blood following an IV bolus is 10ug/ml, the concentration at 1 hour will be 5ug/ml, and 2.5ug/ml at 2 hours. First order kinetics are very common and it is important to understand the ramifications of this mathematical conceptualization of drug elimination. For an example of first order elimination kinetics (Click Here)
First pass effect: Drug that is removed form the circulation by metabolism at the liver. The 'first pass' refers to the fact that orally administered drugs that are absorbed into the circulation, must pass the liver first prior to becoming available to the rest of the body. Thus, drugs that are metabolized in the liver are subject to a large first pass metabolism when administered orally
Fractional occupancy
Free: In receptor binding studies, the free ligand is that which is not bound to the receptor or bound nonspecifically. Often, for the sake of convenience, since the fractional amount bound is so small, the amount added (total) to the assay is taken as the free concentration. However, it is simple to calculate the free concentration by subtracting the bound from the total.
Functional antagonism: see Physiological Antagonism.
G-protein: The linking of many receptors to changes in the cell occurs by activation of a group of heterotrimeric proteins that bind GTP. These proteins are termed Guanine nucleotide binding proteins, or, more simply, G-proteins. The G-proteins are composed of a, b, and g subunits. This group contains many different types ( e.g., Gs, Gi) but they all function by binding GTP. GTP binding to the a subunit will activate signaling. Typically, GTP binding to the a subunit will cause the G-protein to dissociate into a GTP-bound a subunit and a b-g subunit. The a subunit is a GTPase, which hydrolyzes GTP to GDP. This inactivates the signaling process and allows the 3 subunits to reassociate and be available for activation again. (Click Here)Receptors coupled to G-proteins are termed G-protein coupled receptors (GPCR) and often have a 7 transmembrane structure. (Click Here)
G-protein linked receptor: A receptor linked to activation of a G-Protein (see G-protein)
GABA: Gamma-aminobutyric acid. A neurotransmitter with major inhibitory function in the CNS. GABA acts at 2 major types of receptors: the GABA A receptors which are chloride ion channels; and the GABA B receptors which are G-protein coupled.
GFR: see Glomerular Filtration Rate
Gi: The inhibitory G-protein. (see G-protein )
Glomerular Filtration Rate: The GFR is the rate in ml/ min at which filtrate is formed by the kidney glomelurli. Arterial blood enters the glomerlulus and is filtered. The filtrate appears in the urinary tubule. The rate of filtration is typically between 115ml-(females) and 125ml (males) per minute. The rate of filtration can be estimated using a substance (e.g. inulin) that is only filtered and is not reabsorbed or secreted in latter portions of the tubule. Under these conditions, the amount of inulin in the urine will be due only to filtration and thus inulin excretion per minute will be equal to the amount that is filtered by the glomeruli. To determine the rate of filtration, it can be shown that ?
Qe = GFR x Pc ; where, Qe= quantity excreted per minute (mg/min); Pc = plasma inulin concentration (mg/ml).
Thus, GFR=(Qe)/(Pc). Similar estimates can be accomplished using creatinine.
Glycine: One of the neutral amino acid neurotransmitters with significant inhibitory actions in the CNS.
Graded Dose-response
Gs: The stimulatory version of the group of messengers referred to as guanine nucleotide binding proteins. Typically this G-protein acts to increase the conversion of ATP to cAMP by action on the enzyme adenylyl cyclase. (see G-protein )
Half-life: The amount of time for the outcome of a process to increase or decrease by 50%. The half life of a drug is the amount of time required for 50% of the drug to be eliminated.
Hepatic Clearance: The hepatic clearance is the amount of drug removed by the liver as calculated by determining the amount of drug presented to the liver by the hepatic artery and the portal vein and the amount of drug found leaving the liver in the hepatic vein. It should be noted that the liver has the greatest metabolic capacity of any organ. (see clearance)
High affinity binding site
Hill coefficient: The Hill coefficient is derived from the Hill plot which is named after A.V. Hill who developed a method for defining the cooperative binding of oxygen to hemoglobin. Using this method to analyze data, the term nH in the Hill equation takes on values that indicate the nature of binding. When nH = 1, binding is non co-operative and adheres to the law of mass action. Values of nH that differ significantly from 1 suggest a more complex reaction between the ligand and the receptor. When nH ¹ 1, this may be accounted for by multiple classes of binding sites, multistep binding involving several components, or cooperativity. In terms of cooperativity, when nH > 1, positive cooperativity exists; and when nH < 1, negative cooperativity exists. The Hill equation is:
![]()
Hormone:
Hypertension:
Hypnotic:
IC50: The concentration of drug that is required to inhibit the action, binding, or activity of some other drug by 50%. It also is used to describe the concentration of drug that inhibits muscle activity or process by 50%.
ICV: Intracerebroventricular. This abbreviation is often used to refer to an administration route in which drug is administered directly into the cerebral ventricle.
im: Intramuscular. This abbreviation is used to refer to administration of a drug into to a muscle using a hypodermic needle and syringe.
Inhalation: A route of administration in which drug is absorbed via the lung.
Internalization: see Receptor Regulation
Inverse Agonist: This a relatively new term which has been proposed to describe the characteristics of certain drugs that appear to act in a n opposite manner to an agonist at a receptor site. For example, studies suggest that there are inverse agonists at the benzodiazepine-GABA binding site. Unlike agonist which produce an increase in GABA effects, and antagonists which block this action, inverse agonists produce an effect (e.g. anxiety) that is opposite to that produced by the agonists (Anti-anxiety).
Ion trapping
Ionization
ionophore
IP: intraperitoneal. Aroute of administration in which drug is delivered into the peritoneal cavity.
IP3: see diacylglycerol
IPSP: Inhibitory postsynaptic potential
Irreversible antagonist
IT: Intrathecal. Drug is placed in the intrathecal space of the CNS.
IV: An abbreiation for intravenous. A route of administration in which drug is placed directly into the venous circulation. IV administration results in 100% bioavailability and there is no absorption phase to be considered.
K1: the association rate constant. see mass action
K2: the dissociation rate constant. see mass action
Ka : the equilibrium association constant. Ka is the reciprocal of Kd and is a measure of the affinity of a drug for a receptor. It is not the KA, which is equivalent to the Kd
Kd: The equilibrium dissociation constant. It is the concentration of ligand at which 50% of all receptors are occupied. Also used interchangeably with KA. see mass action
Km
Law of Mass Action: see mass action.
LD50 : The dose of a drug that causes death in 50% of the animals tested.
Leukotrienes
Ligand: A ligand is a chemical compound, endogenous or exogenous, that binds to a pharmacologically relevant binding site. The critical pharmacologic feature of a ligand is that it binds; compounds with (agonists) and without (antagonists) intrinsic activity are ligands.
LIGAND: This is a data analysis program developed by Munson and Rodbard (1980) that is most frequently used for binding assays. It employs a nonlinear curve fitting technique to obtain best fitting binding parameters (Bmax, Affinity, Nonspecific binding) for simple and complex binding assays.
Ligand-gated ion channel: A receptor complex in which of a neurotransmitter to the receptor directly causes changes in conductance at an ion channel which is an integral component of the receptor. Examples of this type of channel are the acetylcholine, GABA and glycine receptor-ion channel complexes. Point Here
Lineweaver-Burke Plot: This method is employed to generate a linear function that allows simple estimation of several parameters of receptor binding data. Specifically, the KD and the Bmax (or [LR]max) are determined from a plot of the reciprocal value of the concentration of bound ligand ([B] or [LR]) versus the reciprocal value of the concentration of free ligand [L]. It can also be used for dose-effect data: ED50 and Emax. For binding data, the y-intercept is equal to 1/Bmax and the x-intercept is equal to -1/KD.
The equation for the Lineweaver-Burke plot is:
![]()
The advantage of this linear transformation is that it is simpler to fit a straight line to data, and the maximal binding (or effect) can be estimated from an experiment, even if it was not empirically determined. With the advent of desktop access to nonlinear regression curve fitting programs, this method has been used with less regularity. (Click Here)
Lipophillic
Loading Dose: A loading dose is a relatively large dose of drug that is administered at the start of treatment. The intention is to reach therapeutic steady-state drug levels quickly. Loading doses are usually employed for drugs that reach steady-state levels slowly, or in cases where rapid attainment of steady-state is desirable.
Log dose-response: see dose-response.
Low affinity binding site: A term that is used to describe a binding site for a ligand. This term is a relative descriptor, and only implies that a drug binds with a higher Kd to the low affinity site, compared to some other binding site for the ligand. Ocassionally, low affinity binding site is used to refer to a site for which a drug has a high nanomolar or micromolar affinity.
Macrolides:
Major Tranquilizer: A somewhat antiquated term which was used to refer to drugs for psychoses. E.g. chlorpromazine, haloperidol. see tranquilizer
MAO: monamine oxidase.
Mass Action: The Law of Mass Action most simply stated is that the rate of reaction is proportional to the concentration of reactants. More specifically, the rate of a simple reaction is proportional to the product of the concentrations of the reacting substances each raised to a power equal to the number of moles of that substance in the equation. Therefore, when a chemical reaction is at equilibrium at constant temperature, the product of the active masses on one side of the equation divided by the product of the active masses on the other side of the equation is a constant, independent of the amount of substances at the start of the reaction. Thus, for acids (A) and bases (B):
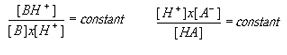

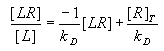
While application of the Law of Mass Action to ligand-receptor interactions
is conceptually useful, it is important to remember that it depends upon several
assumptions. First, the formation of the [LR] complex must be completely
reversible. Second, the ligand and receptor must exist in only 2 states,
bound or unbound. Finally, ligand binds to each receptor with equal affinity
and binding does not alter subsequent affinity.
Michealis Menton Kinetics
Microsomal enzymes
Muscarinic
Nicotinic: A type of cholinergic receptor. The nicotinic receptor is a ligand gated ion channel that responds to acetylcholine. The nicotinic receptor is found on skeletal muscle and postganglionic receptors in the autonomic nervous system.
Nitrates (Syn. organic nitrates): Compounds that are polyol esters of nitric acid. The prototype drug is nitroglycerin which is used to treat angina. Other nitrates include pentaerythritol tetranitrate, erythrityl tetranitrate, isosorbide dinitrate.
Noncompetitive Antagonist: An antagonist that blocks or inhibits the actions of an agonist via its effects at a binding site different from the agonist. A noncompetitive antagonist can be reversible or irreversible. Similarly the effects can be surrmountable or insurrmountable.
Nonlinear Regression: Nonlinear regression uses empirically determined data to estimate a function that describes the relationship between the independent and the dependent variables. The object of fitting the regression is to find the equation parameters that best fit the data. This is accomplished by minimizing the distance of the points (actually the sum of squares) from the fitted line. This is the same goal as for linear regression. However, nonlinear regression requires estimating the initial parameter values, fitting the curve and then repeatedly adjusting the parameter values until the best fit (smallest sums of squares) is found. In a simple case, this might involve estimating a simple hyperbolic fit to one-site binding data:
![]()
In this case, it is necessary to estimate the initial values of KD and BMAX. The repeated adjustment of the parameter estimates is described as an iterative fitting approach, and is typically continued until the sums of squares changes minimally with each further adjustment. At this point the fitting is said to have converged and the parameter estimates are satisfactory. Of course, any data can be fit with any function. Thus, it is important to determine if the curve fit is reasonable. This can be accomplished by determining the goodness of fit which is a measure of the change in the dependent variable which is accounted for by a change in the independent variable. Also the confidence intervals for the parameters can be examined. If they are relatively broad, it is possible that the function does not describe the data well, or that there is significant "noise" in the data.
Nonspecific binding
Normal Distribution: The normal distribution is a common way that the frequency of some value of a variable will be distributed. Simply stated, it is a plot of frequency versus some value (e.g., weight, height, intelligence score). The key parameters of the normal distribution are the mean and the standard deviation. The distribution is bell-shaped and is arrange symmetrically about the mean value for the distribution. The standard deviation is a measure of the spread of the distribution. The normal distribution is also referred to as the Gaussian distribution. (Click Here)
Normal dose-response see dose-response
Operational model of agonism (Black & Leff)
Opiate
Opioid
Oral: The most common and convenient route of administration. However, when drugs are taken by the oral route there are numerous considerations that can affect the actual amount of drug that is absorbed and reaches the site of action. These include the solubility of the drug in the GI system, possible inactivation in the GI tract, the movement of the drug accross biological membranes and the degree of hepatic metabolism (first-pass effect).
Ordered Dose-response
Parasympathetic nervous system The parasympathetic nervous system is part of the autonomic nervous system. This system is a 2 neuron chain in which the first neuron (preganglionic neuron) secretes acetylcholine and the second neuron (postganglionic neuron) also secretes acetylcholine. The parasympathetic division is sometimes refered to as the Craniosacral or cholinergic system. In general, this is a system that is involved with maintenance of bodily activities and conservation of energy (slowing of heart rate, respiration). This system has been characterized as the " rest and digest" system. (Click Here)
Parenteral: Describes routes of administration that bypass the gastrointestinal system. Includes SC, IV, IM, etc.
Partial Agonist: An agonist that does not elicit a full effect, even when bound to all receptors. For an example of the relationship between binding and effect for full and partial agonists (Click Here). Partial agonists may be full agonists for some effects and actually function as antagonists in some cases. Usually as receptor density in a tissue increases the efficacy of a partial agonist also increases. In some cases, a partial agonist can become a full agonist if receptor density increases sufficiently. Similarly, a full agonist may become a partial agonist if receptor density is decreased. Receptor alkylation can easily convert a full agonist into a partial agonist.
Peptides: Two or more amino acids covalently linked by peptide bonds.
Peripheral nervous system: A branch of the nervous system that includes the nervous system outside of the CNS (Brain, spinal cord). It has 2 divisions: the sensory (afferent) and motor (efferent) divisions. The afferent division is involved in conveying information to the CNS. The motor division is divided into the somatic (primarily concerned with voluntary, sketal muscle control) and the autonomic nervous systems (primarily concerned with involuntary regulation of glands, smooth muscle and cardiac muscle). For a diagram (Click Here)
Pharmacodynamics: The branch of pharmacology that deals with the effects produced by a drug.
Pharmacokinetics: The branch of pharmacology that deals with the administration, uptake, distribution and elimination of drugs from the body.
Phase I biotransformation: Phase 1 reactions are often termed as Preparative and include: Hydroxylation, Dealkylation, Sulfoxidation, Reduction and Hydrolysis.
Phase II biotransformation Phase II reactions are often termed as Synthetic and include: Glucoronide conjugation, Sulfate conjugation and Acetylation.
Phosphoinositides: Point here
Physiologic antagonism: When the effect of a drug are reduced or blocked by another drug that produces an opposite physiologic effect. For example, if one drug increases blood pressure, a physiologic antagonist might lower blood pressure not by blocking the receptor effects of the agonists, but by lowering pressure even in the absence of the agonist. Also called functional antagonism.
Placebo:
Postsynaptic receptor: An integral component of a post synaptic cell that acts to convey information to the inside of the cell. The postsynaptic receptor is distinguished from the presynaptic receptor. See receptor.
Potency: Potency is a relative term that compares the ability of drugs to produce a given effect. If one drug can produce the same effect at a lower dose then it is more potent. Technically, potency is determined using dose-response analysis. Comparing the dose-response functions for 2 drugs for the same effect easily allows clear determination of potency. It is not always the case that a more potent drug is more desirable, as it may also be more potent in producing unwanted and bothersome side effects.
Potentiation: When a combination of drugs produces effects that are greater than the additive sum of the effect of each drug.
Presynaptic receptor: A receptor that exists on the axonal side of the synapse. These receptors are often involved in regulating the release of transmitter from the terminal.
Probit: Data can be converted to probits and the probit value plotted against the dose to yield a probit-log dose function. Data that are in percent responding (quantal) form can be easily converted into a probit value using tables. Probits are derived from the normal distribution, where 1 probit unit corresponds to one standard deviation. By convention the mean value is set at 5 and an increase or decrease corresponds to an increase or decrease in one standard deviation. Since the mean value of a normal distribution corresponds to 50%, a probit value of 5 is assigned for the dose that produces a response in 50% of the subjects. Probits are used to linearize data that is normally distributed when plotted against the log of the dose. Linearization can simplify estimating the ED50 and comparison of dose effect curves.
Prostaglandins: Point Here
Protein kinase
Quantal Dose-response: A dose-response function in which responses are determined by the percent of subjects that display a certain experimental endpoint (e.g., death, loss of righting, increase in heart rat of 25bpm).
Radioreceptor Assay
Receptor: Point Here
Receptor Regulation: Click Here
Receptor reserve: see spare receptor
Receptor subtype
Receptor Superfamily
Receptor Tyrosine Kinase: Click Here
Redistribution: A pharmacokinetic term that describes the movement of drug from one comapartment to another. For example, from the vascular compartment to the fat.
Relative potency: Essentially menas the same as potency, since potency is a statement describing the relative doses required for a given effect for 2 drugs.
Renal Clearance: The removal of active drug, or any compound of interest by the kidneys. Renal clearance can be calculated by determining the concentration of drug in plasma flowing to the kidney versus the concentration in plasma leaving the kidney. Clearance is measured in ml/min, which describes the volume of plasma that is cleared of drug per unit time.
Reuptake: When some neurotransmitters are released into the synaptic cleft, a fraction of the release is recovered by transport of the transmitter back into the axon terminal where it may be reused or degraded. This process is called reuptake. Some drugs (e.g., selective serotonin reuptake inhibitors) caninterfere with this process. point here
RIA: RadioImmunoAssay
Rosenthal Plot: see Scatchard Plot.
Routes of administration: The various methods used to introduce a drug into the body so that it reaches the site of action. Includes: intravenous, intramuscular, subcutaneous, oral, rectal, inhalation, transdermal, sublinqual.
Saturation Assay
Saturation Isotherm
Sc: see subcutaneous
Scatchard Plot: In receptor pharmacology the Scatchard Plot provides graphic information on ligand affinity (KD) and the density of receptors (BMAX). The ratio of specifically bound ligand to free ligand is plotted against the bound drug. The slope of the line negative and proportional to the KD (1/ KD), with a steeper line indicative of higher affinity (lower KD). The x-intercept is the estimate of the BMAX. Using the Scatchard Plot, the affinity and receptor density of 2 ligands can be easily compared. For an example of a Scatchard plot (Click Here)
Schild Plot: Click Here
Second messengers: Point Here then see cAMP, cGMP, calcium, IP3, DAG and NO.
Serotonin Receptors: Point Here
Sedative: A drug that reduces agitation or excitement. see tranquilizer
Simple diffusion: The movement of molecules from areas of high to low concentration so as to result in an equilibration of the concentration gradients. Simple diffusion does not require energy expenditure and movement accros a biological membrane can be affected by the charactersitics of a drug (size, charge, lipid solubility).
Spare Receptor: Spare receptors are receptors that do not need to be occupied in order for a maximal drug effect to occur. Limiting the density of spare receptors will not reduce the efficacy of a compound, but will reduce its potency. Spare receptors are drug and tissue specific. In other words, different drugs that act at the same receptor in a particular tissue may have different densities of spare receptors. Furthermore, the same drug at a different tissue site with the same receptor may have more or fewer spare receptors.
Specific Activity: The specific activity of a radiolabelled compound is the measure of the radioactivity per unit mass or volume. This is typically stated in µCi/mg, mCi/mg or Ci/mmol.
Specific binding
Sublingual: Refers to a route of administration in which a drug is retained under the tongue and absorption occurs in the buccal cavity via the mucosa. Venous drainage from the mouth bypasses the liver and empties directly into the vena cava. Thus, drugs susceptibel to a first pass effect will have greater availability when they can be absorbed sublingually. In addition, in cases where a drug passes the biological barriers quickly (e.g., nitroglycerin) onset of drug can be very rapid.
Subcutaneous: A route of administration in which drug is deposited under the skin using a hypodermic needle.
Symapthetic nervous system: The sympathetic nervous system is part of the autonomic nervous system. This system is a 2 neuron chain in which the first neuron (preganglionic neuron) secretes acetylcholine and the second neuron (postganglionic neuron) secretes norepinephrine, or in the case of the adrenal medulla, epinephrine. The sympathetic division is sometimes refered to as the thoracolumbar or adrenergic system. In general, this is a system that is involved with activation of bodily activities and mobilization of energy consuming activities (increase in heart rate, respiration). This system has been characterized as the " flight or fight" system. (Click Here)
Sympathomimetic: Drugs which have effects similar to epinephrine and norepinephrine. These drugs tend to have activating effects on the sympathetic nervous system.
Synapse: Consists of the functional unit of neurotransmission. Includes the presynaptic element, the synaptic cleft and the postsynaptic element.
Synaptosome: These are preparations from nervous tissue that consist of broken-off (or pinched-off) nerve endings. The synaptosomal preparation is made by homogenizing tissue followed several centrigugation steps. The synaptosome typically includes a portion of the presynaptic terminal and the adjacent post-synaptic membrane. The presynaptic portion of the synaptosome usually reseals and can form a funtional terminal containing mitochondria, transmitters, vesicles and enzymes.
Tachyphylaxis: A rapidly developing desensitization or tolerance to a drug. Often this is used to describe decreased response to a drug that occurs soon after administration when active drug levels might imply that sufficient drug is available to induce a full response.
Thromboxanes Eicosanoids derived from the cyclooxygenase pathway. The thromboxanes have effects on a variety of tissues including: smooth muscle (especially bronchial and vascular) and platelets. Thromboxane A2 produces platelet aggregation and vasoconstriction and contraction of bronchial smooth muscle.
Tolerance: Following exposure to a drug, there is a reduction in the response to subsequent administration of the drug. Tolerance can be empirically defined as a shift to the right in the does-response curve with an associated increase in the ED50 or EC50. There may also be a reduction in the maximal effect (Emax).
Topical: see transdermal
Toxicity: The ability of a drug or chemicalm to cause injury or adverse effects.
Tranquilizer: A relatively outdated term that refers to drugs that bring about sedation, quieting, reduce anxiety and agitation. Tranquilizing drugs have been classified as major and minor tranquilizers. The major tranquilizers are employed for severe psychiatric disorders such as psychoses, and the minor tranquilizers are used to treat less severe disorders such as insomnia and anxiety.
Transdermal: Point Here
Upregulation: This term is often used to refer to an increase in neurotransmitter receptor density. An increase in receptor density can lead to increased potency of drugs acting at that receptor, and can increase the maximal effect of partial agonists. In some cases the activity of drugs that are extremely weak can be substantially enhanced by upregulation. see receptor regulation
Uptake
Vasodilators: A group of drugs that are used for the treatment of heart failure, angina and hypertension. This group includes drugs with diverse mechanisms of action including the organic nitrates, calcium channel blockers, a blockers, "direct" vasodilators (e.g., hydralazine), ACE inhibitors and antagonists, and others. Overall, these drugs appear to dilate the peripheral vasculature either on the venous or arterial side, or both.
Vesicle: A component of the nerve (axon) terminal in which neurotransmitters are believed to be stored prior to release. Release occurs when an action potential arrives at the nerve terminal and through a series of events causes the vesicles to fuse with the cell wall, open up and release their contents in to the synaptic cleft.
Volume of distribution (Vd): This is a hypothetical number expressed in liters which provides information on the degree to which a drug is distributed. The Vd is the ratio of the absolute amount of drug (e.g. mg) in the body to the concentration (e.g. mg/ml) as typically determined in plasma. If a drug (100mg) is administered i.v. and its Vd (100L) is large then its concentration in plasma is low (e.g. .001mg/ml) and it is widely distributed out of the plasma water compartment. If the concentration is relatively high (0.1mg/ml) then the Vd is low (10L) then the drug is retained in the plasma compartment. That the Vd can take on values that are clearly hypothetical is made clear by considering that Vd values can range far in excess of the actual fluid volume of a normal individual. For an example of Volume of Distribution (Click Here)
Withdrawal Syndrome: Point Here
Comments?? Suggestions?? Please send them to me at YoburnB@StJohns.edu
Or use the form below:
![]()
This page has been accessed
times
Since September 23, 2000
Updated
April 10, 2003
![]()
This material is the property of:
© Byron C. Yoburn, Ph.D.
College of Pharmacy, St. John's University, Queens, NY 11439
Unauthorized use is prohibited
![]()
{kind=link}
{kind=link}